MassChroQ (Mass Chromatogram Quantification) is a fast and versatile software that performs alignment, XIC extraction, peak detection and quantification on data obtained from Liquid Chromatography-Mass Spectrometry (LC-MS) techniques.
A full description and evaluation of MassChroQ have been published in:
MassChroQ is able to:
- analyze data obtained from both high and low resolution spectrometer systems;
- analyze label-free as well as isotopic labeled data (e.g. SILAC, ICAT, N-15, C-13, etc.);
- take into account complex data treatments as peptide or protein fractionation prior to LC-MS analysis (SCX, SDS-PAGE, etc.);
- time-efficiently process a large amount of differently analyzed samples in one shot (by grouping similar samples and offering the possibility to parametrize the analysis of each group).
Moreover, in MassChroQ:
- every analysis step (signal filtering, XIC extraction, peak detection, alignment, grouping of samples) is fully configurable, so that the user can adapt every parameter to best fit its data;
- every analysis step (signal filtering, XIC extraction, peak detection, alignment) produces precise traces, so that the user can easily follow and check their relevance and also adjust its analysis parameters if necessary;
- every used and produced data is in open standard format (mzXML, mzML, masschroqML, gnumeric, csv, …).
MassChroQ graphical user interface
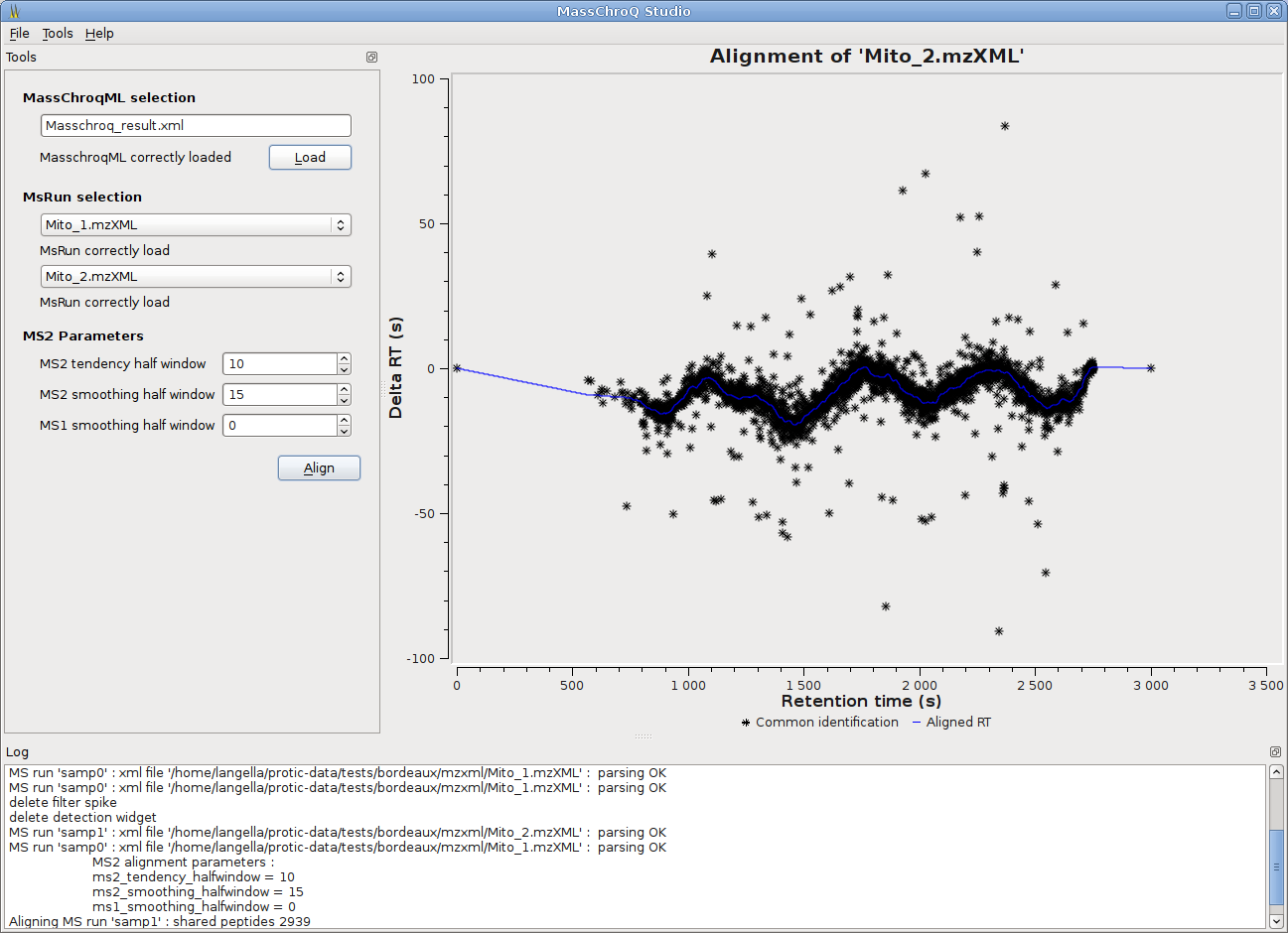
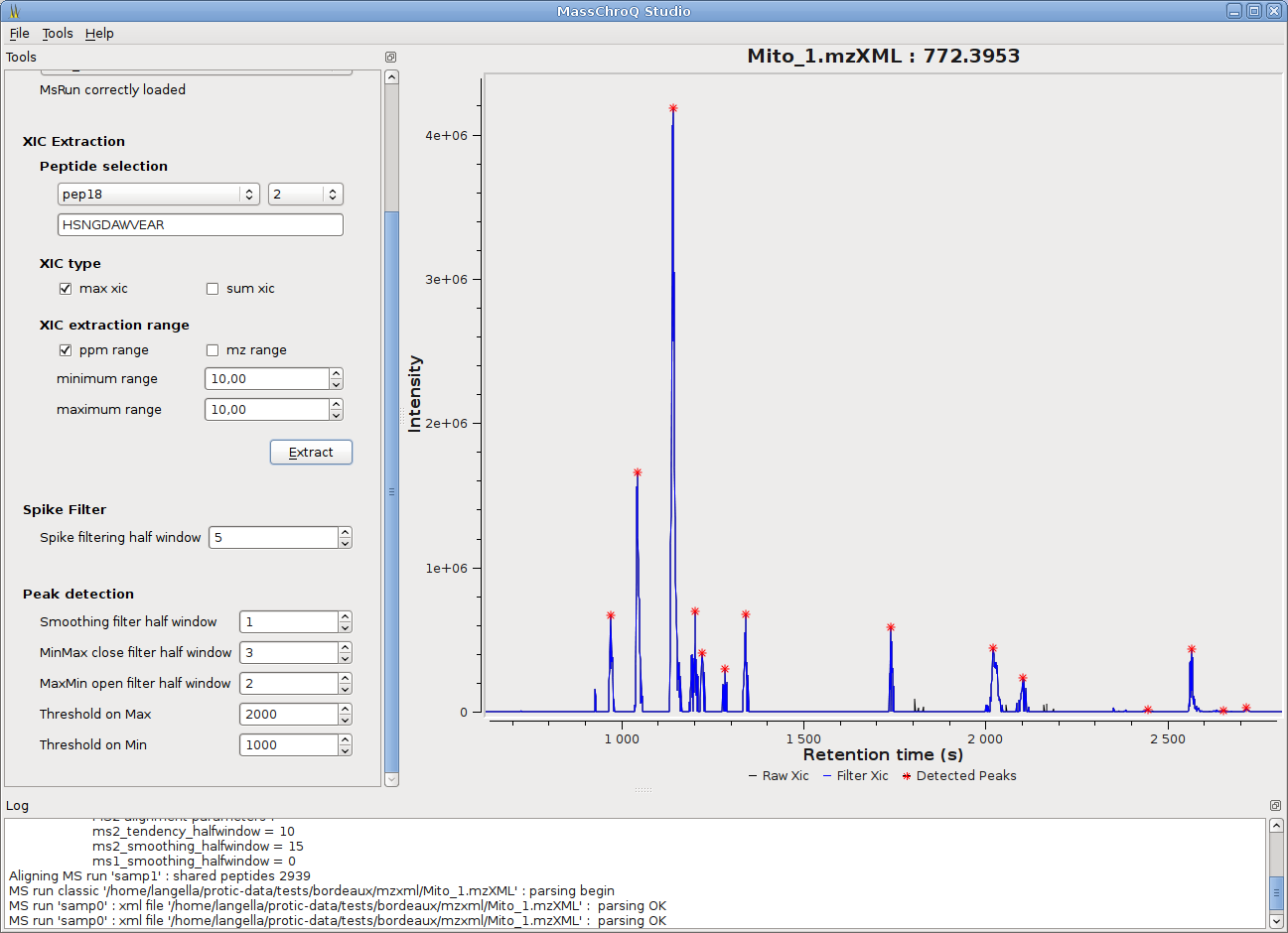
MassChroQ has now a gui to seamlessly run MassChroQ ML files, and graphical tools to visualize extracted ion chromatograms (XICs), detected peaks, alignments between runs…
MassChroQ main features
- Parsing of mzXML and mzML LC/MS data formats.
- Quantification of all the identified peptides of the samples and/or a given list of mass over charge (m/z) values and/or a given list of (m/z, retention time) pairs of values.
- The identified peptides can be automatically integrated into MassChroQ’s analysis by using our X!Tandempipeline (which performs identification and exports the results in masschroqML format) or via TSV (Tab Separated Values) files containing the identification results obtained from other identification software.
- Two alignment methods are available: the third-party OBI-Warp alignment based on MS level 1 data is integrated in MassChroQ, and the in-house developed MS2 alignment based on MS level 2 information.
- The quantification in MassChroQ is performed on XICs (eXtracted Ion Chromatograms): after XIC extraction and filtering, peaks are detected on them by using morphological transforms (see the user manual for details). Peak areas (i.e. quantification value) and boundaries are then computed.
- Peak matching is performed (after alignment if any) as follows: a detected peak is assigned to an identified peptide if and only if the (aligned) retention time of this peptide is located within the peak boundaries. Peak matching in MassChroQ can be performed in two rounds: a first one during quantification, and once quantification is finished, the user can perform a second, improved peak matching round, taking into account the retention times of the previously matched peaks.
- Various XIC filters are available : moving median/mean filters to smooth the signal, background and baseline noise removal filters, anti-spike filter to remove spikes caused by some high resolution spectrometers, morphological filters, etc.
- The user can export results in tsv, gnumeric, xhtml and masschroqML formats. They are ready for automatical use in statistical software (like R) or for manual/visual exploration.
- Evaluation of MassChroQ on complex label-free data obtained from both low and high resolution mass spectrometers allowed the measurement of low CVs for technical reproducibility (1.4%) and high coefficients of correlation to protein quantity (0.98) (Valot, Langella, Nano, Zivy 2011).
- Two quantitative studies to be published soon, one label-free and the other containing dimethyl labeled data together with SCX-fractionation (pubmed), have shown the accuracy and precision of MassChroQ’s quantification.
License
You do not have to pay or ask for permission to use MassChroQ, because it is Free Software (free as in Freedom) and freely available (free as in beer) under the GNU General Public Licence version 3. Note, however, that if you like MassChroQ and you use it for your research, you have to cite our work. You can also take a deeper look into the Free Software philosophy that MassChroQ supports, for example from here or from here.
Releases
The current release version of MassChroQ is version called “ ”. We think that this release version is good for production use. Windows binaries and Linux packages can be downloaded from the MassChroQ Download page.
Documentation and Help
Documentation about MassChroQ, including the User Manual, can be found on the MassChroQ Documentation page. An FAQ is available here. The main mailing list for news, user questions and help about MassChroQ is the pappso-tools mailing list.
MassChroQ community tools
Community tools (bug tracker, user forums) and the git source code repository for MassChroQ are hosted on the MassChroQ page at ForgeMIA.
- MassChroQ’s Git repository can be checked out with anonymous access using the following command:
git clone git@forgemia.inra.fr:pappso/masschroq.git
- The bug tracking system is available here.
MassChroQ is a Free and Open Source project and everyone is very welcome to participate, collaborate or simply discuss with us about it.
Acknowledgments
The authors would like to thank:
- INRAE ForgeMIA, for effectively hosting the MassChroQ project.
- Mélisande Blein-Nicolas, for her participation in the MS2 alignment algorithm conception and in testing and improving the not-stable-at-all first private versions of MassChroQ.
- Ludovic Bonhomme, not only for his crucial guinea pig role in the testing and improving phases of MassChroQ, but also for taking revenge by turning MassChroQ into a guinea pig for his research. We are sure that he will particularly appreciate version 2.0 betas coming soon.
- The open source OBI-Warp alignment library that we use in MassChroQ, and its author, John T. Prince, for his collaboration.
- IBiSA, for their financial support to the MassChroQ project.