i2MassChroQ is a free software (GPL v3) that helps the user
- filter and group peptide/protein identifications from MS/MS mass spectra;
- perform area-under-the-peak XIC-based quantification of proteins.
i2MassChroQ is fully described in:
Langella O, Renne T, Balliau T, Davanture M, Brehmer S, Zivy M, Blein-Nicolas M, Rusconi F.
(2024-08-02)
Full Native timsTOF PASEF-Enabled Quantitative Proteomics with the i2MassChroQ Software Package.
J. Proteome Res.,
8
(23)
3353-3366
The rewrite in C++ has brought major performance improvements along with a host of new features.
Main features
- Easy to install and run (Windows 64bits or Linux)
- Reads SAGE json and TSV results files
- Reads X!Tandem xml results files
- Reads MASCOT dat results files
- Reads TPP pepXML results files
- Reads PSI mzIdentML results files
- Offers a graphical user interface (gui) to run X!Tandem analyses
- Various filters based on statistical values at peptide and protein levels
- Powerful original grouping algorithm to filter redundancy
- “phosphopeptide” mode to handle phosphoproteomic datasets
- Edit, search and sort your data graphically
- XIC browser (eXtracted Ion Chromatogram)
- Comparisons of theoretical isotope patterns to measured MS1 XIC areas
- Exports your data directly to Microsoft Office 2010 and LibreOffice (ods export)
- Hyperfast huge dataset processing quickly
- Peptide quantification through MassChroQml export
- Online support through the pappso-tools mailing list
Graphical user interface
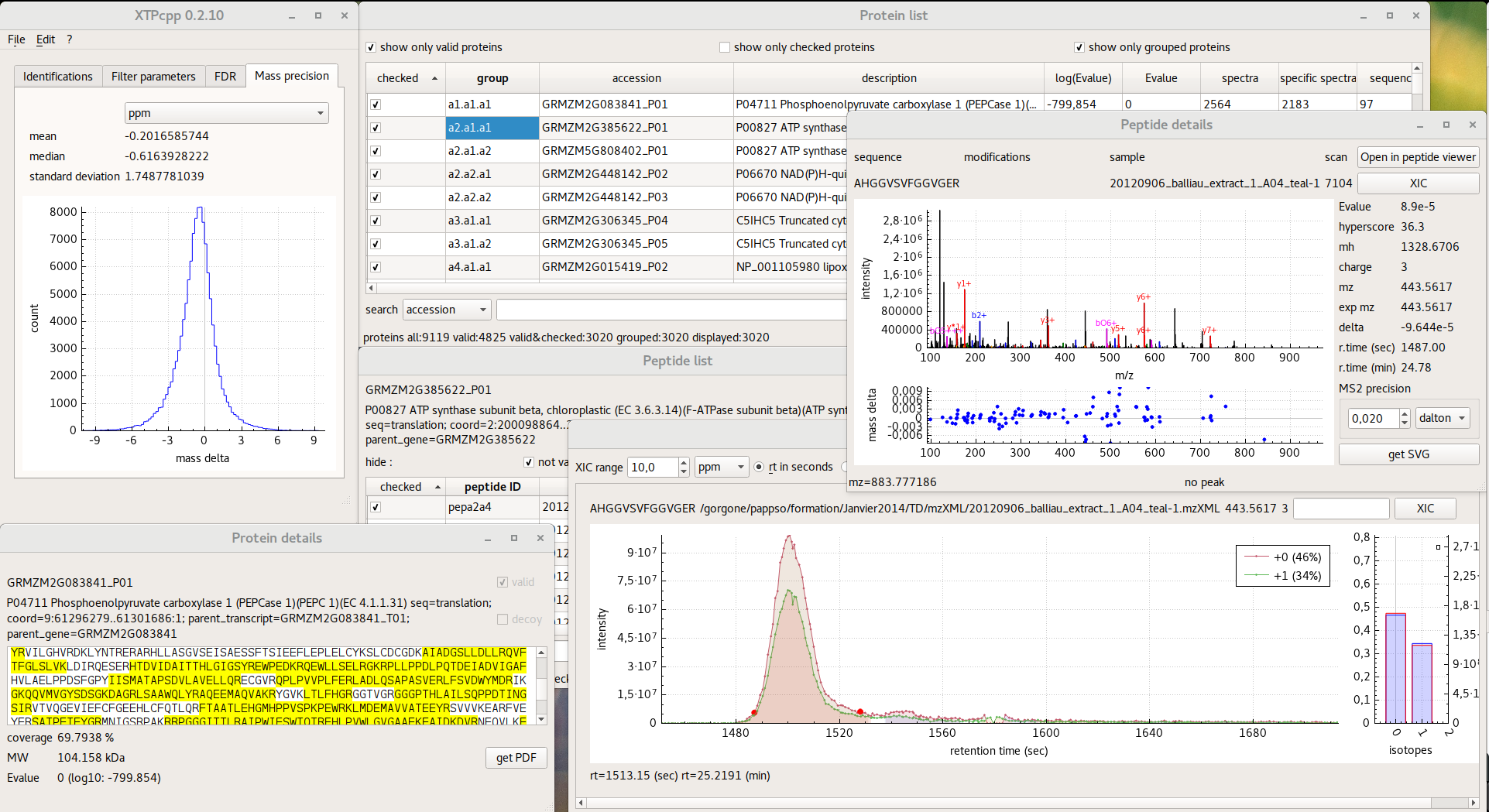
Grouping algorithm
Our grouping algorithm removes non-informative redundant information from the dataset and builds clusters of proteins (groups and subgroups). Redundancy of protein databases is fully filtered as follows:
- Proteins identified without specific peptides compared to others are eliminated
- Proteins identified with the same pool of peptides are assembled (subgroups)
- Proteins are grouped by function (identified with at least one common peptide), and the specific peptides for each sub-group of proteins are highlighted
Performances
i2MassChroq was developped to be fast, with a low memory footprint.
For example, given a common dataset :
- 12 complex biological samples (iPRG 2015 study)
- corresponding to 2619 identified proteins, 24548 unique peptides
- and 193657 MS/MS spectra
would give on a basic computer (1,4Go RAM, 3Ghz CPU) :
- 2419 groups of proteins (same protein function, at least one common peptide)
- 2586 subgroups of proteins (each protein sharing the same set of peptides)
- 59 seconds total runtime to read, filter and group the 12 MS identification files (X!Tandem XML files)
- 1 second only to filter and group
Acknowledgments
Many thanks to the Global Proteome Machine organisation (the GPM) that develops the X!Tandem free search engine software. Many thanks to ForgeMIA, for effectively hosting the i2MassChroq project.