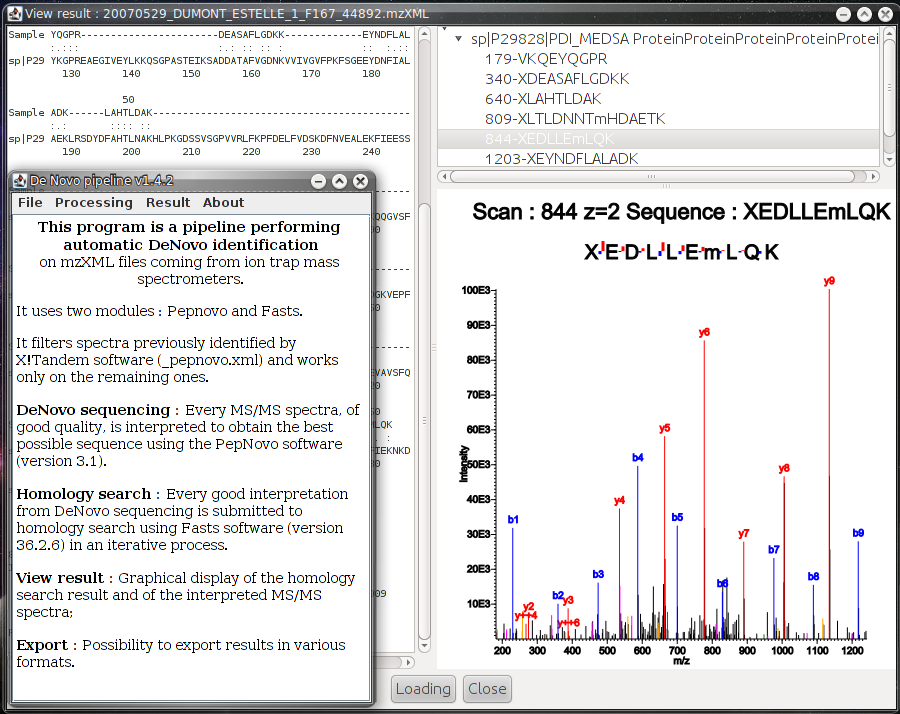
The classical method for protein identification of LC-MS/MS data uses database searching and matching, as it is the case in Mascot, Sequest or X!Tandem softwares. However, these identifications are possible only if the protein being searched is present in the database. To address this problem the de novo interpretation strategy can be used instead. There exist softwares that use this strategy, but they are often difficult to use in a direct, automated way.
The De Novo Pipeline uses the de novo technique to perform identification on data collected from ion trap mass spectrometers. It performs automated analysis by connection of two applications:
- PepNovo: automated interpretation of MS/MS spectra in a possible peptide sequence,
- Fasts: homology search in an iterative mode, to identify proteins from peptides sequences.
The De Novo Pipeline is complementary to X!Tandempipeline: it allows you to remove spectra previously identified by X!Tandem and perform analysis on the remaining ones. The results can be graphically viewed and/or exported into tabulated files. Installation and running procedures are explained in the documentation.
This application requires java 1.6, which you can get here.