Voici les changements depuis la dernière version annoncée 0.4.27 :
- Meilleure prise en charge des données du timsTOF pro : amélioration des performances, visualisation et export de nouvelles données (fenêtre de mobilité 1/K0, énergie de collision)
- Intégration de MassChroQ 2.4.3, prise en charge du timsTOF pro pour la quantification des peptides.
- Identification de peptides avec X!Tandem directement sur les données du timsTOF.
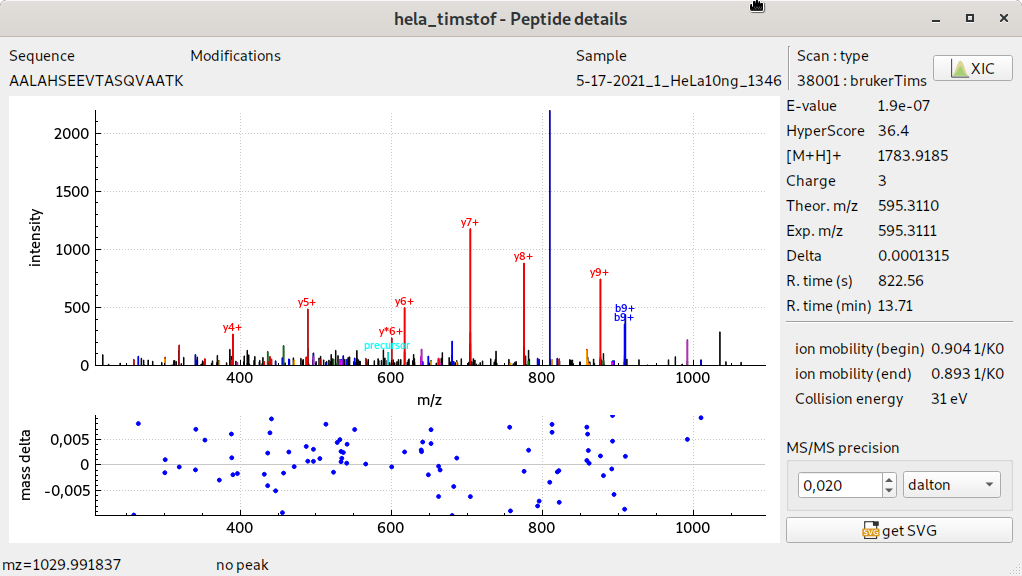
visualisation de spectres MS/MS timsTOF pro
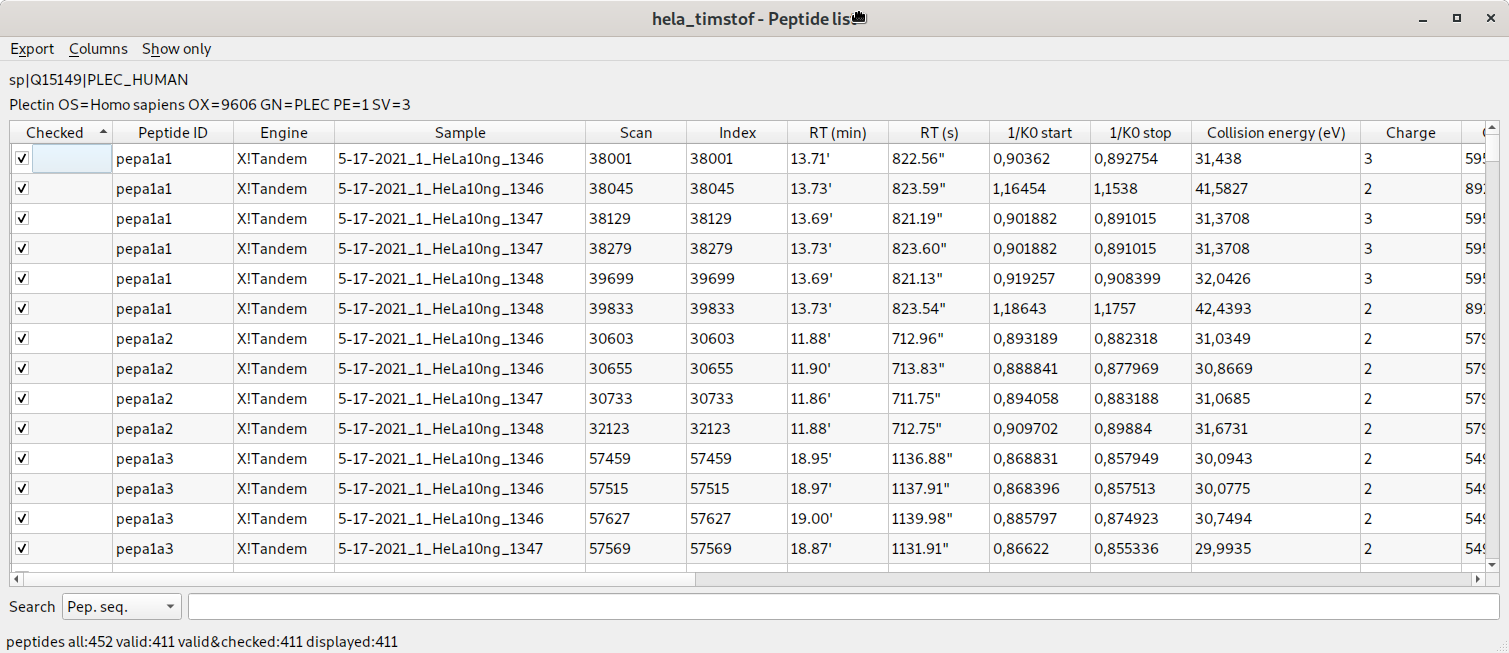
liste des peptides identifiés contenant les informations de mobilité ionique
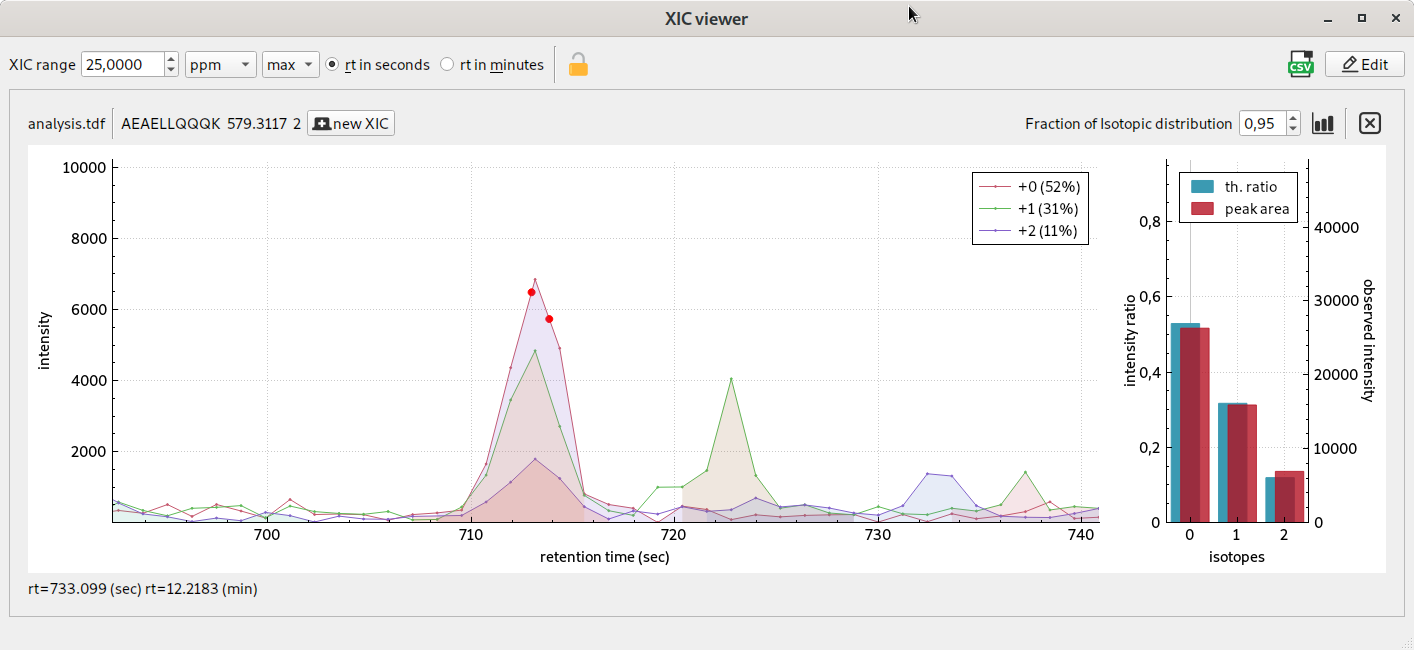
visualisation de XICs, courant d’ion extrait dans la plage de mobilité
Merci beaucoup à toutes l’équipe PAPPSO pour ces améliorations et notamment à :
- Filippo Rusconi, pour le développpement en C++ et la documentation.
- Thomas Renne, apprenti en bioinformatique, qui a développé la plupart des nouvelles fonctionnalités pendant ses 2 dernières années.
Un nouveau module de post traitement des intensités de peptides sera bientôt disponible pour faciliter la reconstruction des abondances de protéines à partir des intensités de peptides, visualisation/exploration des données, filtration, imputation des données, analyse des variations d’abondances pour différents scénarios : labdel free, séparation par fractionnement (SDS), marquage isotopique.
Vous pouvez participer au développement, proposer de nouvelles fonctionnalités ou signaler un bug depuis : ForgeMIA gitlab