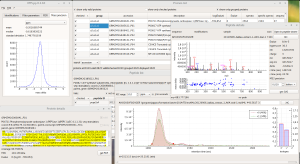
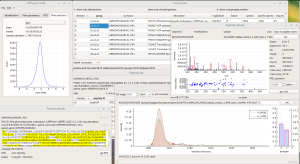
ALL-P is a statistical framework based on a hierarchical modeling that takes into account shared peptide information for estimating protein abundances. ALL-P performs a simultaneous analysis of all the quantified peptides, handling the biological and technical errors as well as the peptide effect.
Compared to a method based on the analysis of one protein at a time that does not include shared peptides (ONE-P method), ALL-P proved to be far more reliable for estimating protein abundances and testing abundance changes.