X!TandemPipeline C++ version 0.4.42
Voici les changements depuis la dernière version annoncée 0.4.27 :
- Meilleure prise en charge des données du timsTOF pro : amélioration des performances, visualisation et export de nouvelles données (fenêtre de mobilité 1/K0, énergie de collision)
- Intégration de MassChroQ 2.4.3, prise en charge du timsTOF pro pour la quantification des peptides.
- Identification de peptides avec X!Tandem directement sur les données du timsTOF.
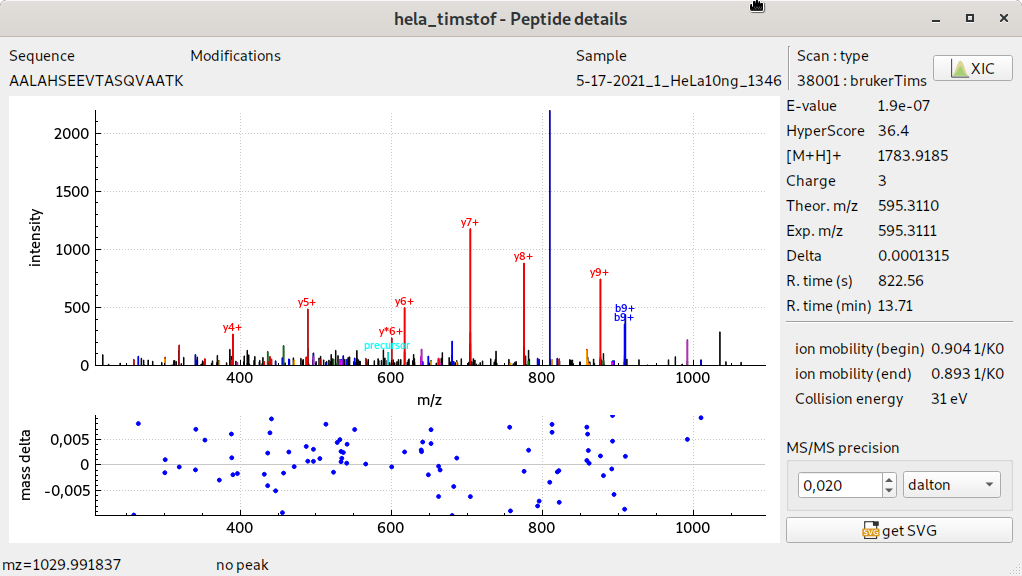
visualisation de spectres MS/MS timsTOF pro

