i2MassChroQ User Manual
- Preface
- 1 Generalities
- 2 Fundamentals in Bottom-up Proteomics
- 3 The main program window
- 4 Exploring identification data
- 5 Exploring post-translational modification data
- 6 Advanced Proteomics Configurations
- 7 i2MassChroQ and Quantitative Proteomics
- 8 Specific procedures for the timsTOF line of instruments
- A GNU General Public License version 3
8 Specific procedures for the timsTOF line of instruments
This chapter describes the very few specific procedures to carry out when the proteomics data at hand are from the Bruker timsTOF line of instruments.
8.1 General considerations #
The mass spectrometers from the timsTOF line of instruments by Bruker implement ion mobility mass spectrometry by trapping ions and subjecting them to a gas flow that moves them in the trap according to their collisional cross section. Interestingly, the outcome of that operation is the reverse of conventional drift tube-based ion mobility: large ions are released first and smaller ions are released last.
Apart from the observation above, the result is nonetheless that ions entering the instrument are separated according to a new dimension that is orthogonal to the other two retention time and m/z ratio dimensions: the ion mobility dimension. This new dimension inevitably introduces more complexity and greater volume to the mass data.
In a historic move, Bruker has decided to publish the technical details of
their data format. During the acquisition, data are stored in two separate
files located in their data directory that has the .d extension:
analysis.tdf: this file is a SQLite3 relational database that contains all the metadata about the acquisition. The generic metadata term defines data that describe the actual data. So this file contains data that explain how the data are organized in the actual data file below.analysis.tdf_bin: this file is a binary-format file that holds the data in the form of a succession of numbers packed according to a specific scheme that Bruker has decided to make public.
The SQLite3 analysis.tdf
file contains a set of tables. Most often, records in one table make
reference (relate) to other record(s) in other
table(s). This is why this database file is said to be a
relational database. A view of the tables making that
relational database file is shown in Figure 8.1, “View of the relational database file”.
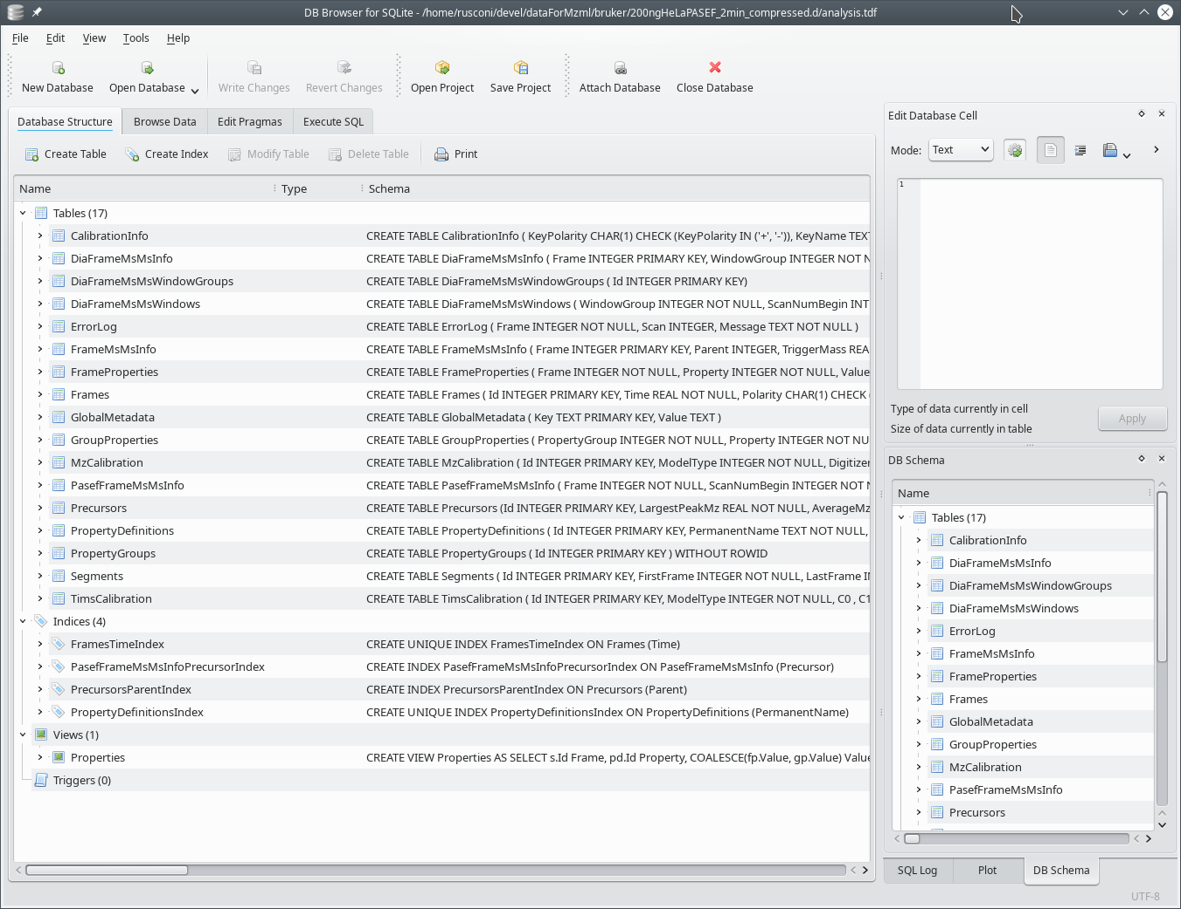
The mass spectrometers of the timsTOF line of instruments by Bruker
produce mass data that are stored in two files. This figure shows
the table structure of the relational database
analysis.tdf file displayed in
SqliteBrowser (a Free Software
application).
Figure 8.1: View of the relational database file #
When dealing with proteomics projects that have their data originating in timsTOF instruments, some specific steps are to be taken so as to inform i2MassChroQ that specific handling is required. These will be reviewed below in the same succession as they need be implemented when running i2MassChroQ.
8.1.1 Running X!Tandem identifications with Bruker timsTOF data #
It is possible to load Bruker timsTOF data right in the i2MassChroQ program's graphical user interface, as shown in the window pictured in Figure 3.2, “X!Tandem-based identification configuration”. The very first specific step to take in this case is to select the data files by clicking the Add Bruker timsTOF folders.
The Add Bruker timsTOF folders button lets the user
choose the Bruker data directory (.d extension) and then asks if the data to be
loaded are in the TDF or MGF format (see Figure 8.2, “File format selection dialog for Bruker timsTOF data”).
Indeed, the MGF file generated by the Bruker software is automatically
installed in the .d extension
data directory.

When handling Bruker timsTOF data, two file formats are available: MGF and TDF. See text for details.
Figure 8.2: File format selection dialog for Bruker timsTOF data #
The DataAnalysis software from Bruker allows one to export proteomics MS/MS data into MGF format files (Mascot generic format). Their native data format, though, is the TDF format. It is important to keep in mind that the MGF format only stores MS/MS spectral data, no MS data. By using this format, i2MassChroQ and MassChroQ won't be able to access MS data, which are required in a number of situations, in particular when extracting ion currents for given m/z ratios, for example, or for area-under-the-curve quantitative proteomics. It is thus always recommended to use the native TDF format whenever available.
When loading Bruker timsTOF data right into i2MassChroQ as described above, the software performs some under-the-hood operations that the user might want to be aware of. The hidden operations are unveiled in the following sections, as they involve command line programs shipped along with i2MassChroQ that might be of interest to the user.
8.1.2 Converting Bruker timsTOF data to mzXML with mzxmlconverter #
X!Tandem needs the mass spectrometric data that it uses for the database searches to be in the mzXML format. For this very reason, i2MassChroQ cannot work by harnessing the capabilities of X!Tandem starting from Bruker timsTOF data. These data need to be converted to an mzXML file before they can then be fed to X!Tandem.
In order to be able to store a mzXML file on disk, the user may convert timsTOF data files (TDF or MGF) to mzXML using the mzxmlconverter program that is shipped along with i2MassChroQ. This program is a command line program that takes a data file in input and that writes a mzXML file in output. The command line syntax is easy, as picture in Figure 8.3, “Converting mass data files to mzXML data files”.
To obtain help about the program, run the following:
$
[16]
<path_to>/mzxmlconverter --help RETURN
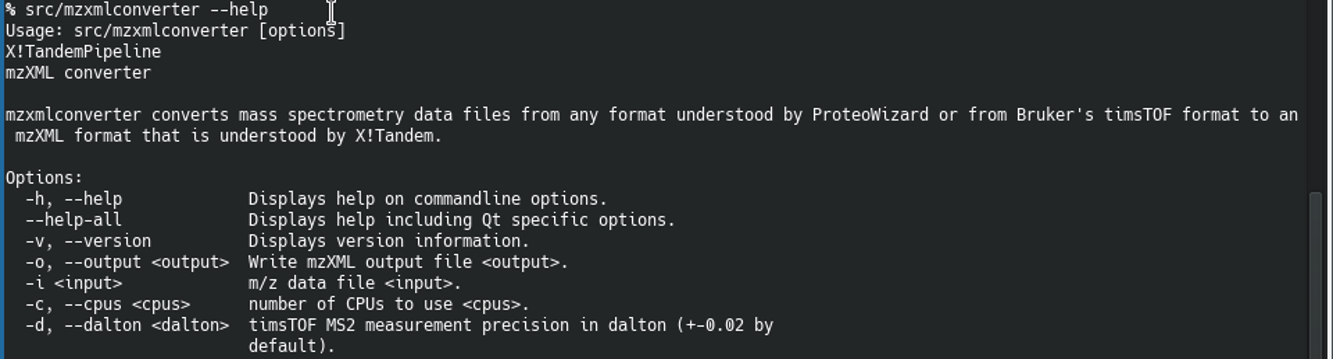
Files of any format handled by ProteoWizard or files from the Bruker's timsTOF line of instruments can be converted to mzXML using mzxmlconverter. Conversion from timsTOF format to mzXML is performed entirely by our own software.
Figure 8.3: Converting mass data files to mzXML data files #
Warning: The mzXML file format does not contain mobility data from the timsTOF data files
It is important to grasp that the mzXML file that is generated by mzxmlconverter does not contain all the mass data that are contained in the Bruker's timsTOF data files. When loading the produced mzXML format files in i2MassChroQ, the X!Tandem program will be able to perform peptide and protein identifications. Later, however, when the user will try to activate features in i2MassChroQ that require the original data, the software will not be able to provide the expected results, like XIC reports or ion mobility values, because it won't have access to the original data.
The mzxmlconverter program is practical because it allows storing the mzXML file on disk and loading it in i2MassChroQ for X!Tandem to consume it for the identifications. However, as stated in the warning above, that mzXML file has not a full copy of the data in the original mass spectrometry data file (be that a mzML, or MGF, or TDF file). i2MassChroQ has a solution for this problem: using an integrated workflow to convert the original data file to mzXML, make X!Tandem use it, write out the X!Tandem results file and finally rewrite that file into a new version by adding a connection between the X!Tandem run results and the original mass data file. In this way, when the user will activate features in i2MassChroQ that need accessing the original mass data file, the expected results will be effectively displayed. This process is described in detail in the next section.
8.1.3 Data conversion process with Bruker timsTOF TDF data and tandemwrapper #
In order to seamlessly use Bruker timsTOF data in the context of performing X!Tandem identifications and later MassChroQ-based quantifications, the tandemwrapper program is made available to users who want to perform database searches using the command line interface. The tandemwrapper program performs an under-the-hood file format conversion as described in the previous section before automatically feeding the generated mzXML file to X!Tandem. After X!Tandem has produced its results file, that file is rewritten by tandemwrapper in such a manner that the connection with the original mass spectrometry data files is reinstated for further use in the i2MassChroQ graphical interface.
There is, however, a way to convert Bruker timsTOF data files to mzXML using a standalone program, called tandemwrapper, that is shipped along with i2MassChroQ. The tandemwrapper program is a command line program that takes as input a XML configuration file. The XML file is most similar to the configuration file that X!Tandem uses.
To obtain help about the program, run the following:
$
<path_to>/tandemwrapper --help RETURN
A typical tandemwrapper input configuration file is shown below:
<?xml version="1.0" encoding="UTF-8"?>
<bioml label="example-tandemwrapper-mass-data-file.mzxml">
<note type="heading">Paths</note>
<note type="input" label="list path, default parameters">/full_path_to/xtandem-presets-file.xml</note>
<note type="input" label="list path, taxonomy information">/full_path_to/database.xml</note>
<note type="input" label="spectrum, path">full_path_to/mass-data-file.mzxml</note>
<note type="heading">Protein general</note>
<note type="input" label="protein, taxon">usedefined</note>
<note type="heading">Output</note>
<note type="input" label="output, path">/full_path_to/tandemwrapper-output.xml</note>
</bioml>
The XML configuration file that is provided to tandemwrapper on the command line is a replicate of the file that X!Tandem itself expects. That file is shown in Figure 8.4, “Configuration file for tandemwrapper”.
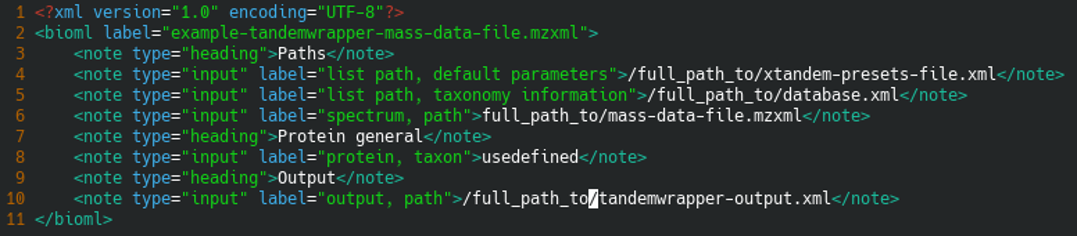
The tandemwrapper program takes as input a configuration file that is most similar to the configuration file that is fed to X!Tandem.
Figure 8.4: Configuration file for tandemwrapper #
The following elements need an explanation:
default parameters: In the example, the
/full_path_to/xtandem-presets-file.xmlfile is the X!Tandem presets file, already discussed in Section 3.3, “Setting the X!Tandem Run Presets”.taxonomy information: In the example, the
/full_path_to/database.xmlfile is the file that configures the location of the FASTA protein database files that are searched by X!Tandem. This file is described below.path: In the example, the
/full_path_to/mass-data-file.mzxmlfile is the mass spectrometry data file in the mzXML format.
Tip
The mass spectrometry data file might be of any format that can be handled by ProteoWizard (open data formats only, particularly mzML) and also the Bruker's timsTOF TDF format that is handled by our own code.
output, path: In the example, the
/full_path_to/tandemwrapper-output.xmlfile is the file in which the X!Tandem configuration file is written for immediate use by X!Tandem.
The configuration file that indicates where the FASTA protein database files are located, that is referenced in the tandemwrapper input configuration file is shown in Figure 8.5, “Configuration file pointing at the FASTA protein databases”.
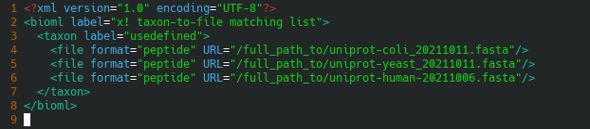
This file tells tandemwrapper the location of the FASTA protein databases required when X!Tandem will actually perform the searches.
Figure 8.5: Configuration file pointing at the FASTA protein databases #
The tandwrapper program performs the following tasks in sequence:
Convert the input mass data file to the mzXML file format that X!Tandem needs to perform the database searches. This step is only performed if the original mass spectrometry data file has not the mzXML format. X!Tandem produces an identification results file in an XML format;
The mzXML format that is consumed by X!Tandem is a pretty simple format that was not designed to store a large variety of data/metadata, like ion mobility data, for example. For this very reason, tandemwrapper reads the identification results file produced by X!Tandem (that file is also in a specific XML format) and rewrites it to a new analogous file that has all the necessary connections to the original mass data file. In this way, when the new version of the X!Tandem identification results file is loaded in i2MassChroQ all the original mass data can be accessed to provide the user with all the data, like XIC chromatograms, ion mobility data, for example. To load the X!Tandem identification results, see Section 3.4, “Loading the Protein Identification Results”.