i2MassChroQ User Manual
- Preface
- 1 Generalities
- 2 Fundamentals in Bottom-up Proteomics
- 3 The main program window
- 4 Exploring identification data
- 5 Exploring post-translational modification data
- 6 Advanced Proteomics Configurations
- 7 i2MassChroQ and Quantitative Proteomics
- 8 Specific procedures for the timsTOF line of instruments
- A GNU General Public License version 3
6 Advanced Proteomics Configurations
This chapter describes in detail all the various advanced configurations that determine the workings of i2MassChroQ in various contexts.
Some of the advanced proteomics configurations can only be carried over when protein identification results have been loaded in i2MassChroQ. Indeed, only at that point, does the menu of the main program window show the submenus of interest, as shown in Figure 6.1, “The menu showing various configuration tasks”.
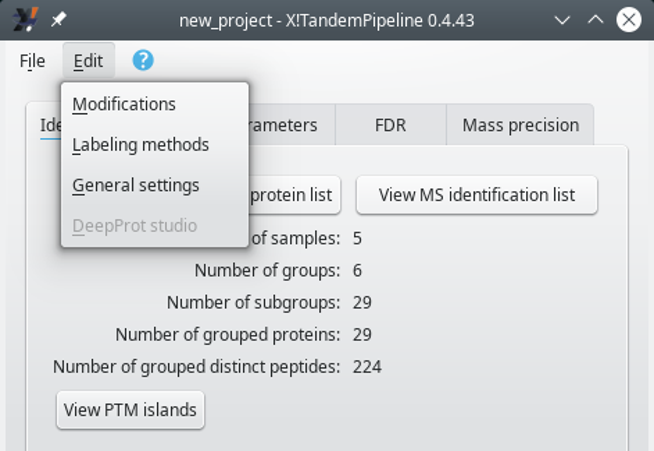
This menu becomes available in the main program window only when identification data have been loaded in i2MassChroQ
Figure 6.1: The menu showing various configuration tasks #
6.1 Configuring Modifications #
It is often required to specify the chemistry of a given chemical modification that might be performed (or spontaneously observed ) on the proteins of a sample. For example, proteins in a sample often undergo a chemical modification of the cysteine residues in their reduced form to block them from reoxidizing into a sulphide bond. The reagent used is iodoacetamide and the reaction name is carbamidomethylation.
In order to be able to access the modifications configuration, select from the menu. The window that opens up is shown in Figure 6.2, “Modifications editor window (folded)”. In that Modifications editor window, the currently defined modifications are listed with no details about them.
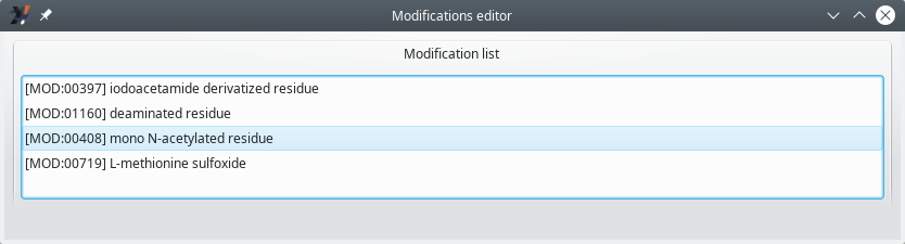
When the Modifications editor window is displayed, all the modifications data are folded.
Figure 6.2: Modifications editor window (folded) #
To start editing an existing modification, click the correponding item from the list, which opens up a Selected modification group box widget, as shown in Figure 6.3, “Modifications editor window (unfolded)”.
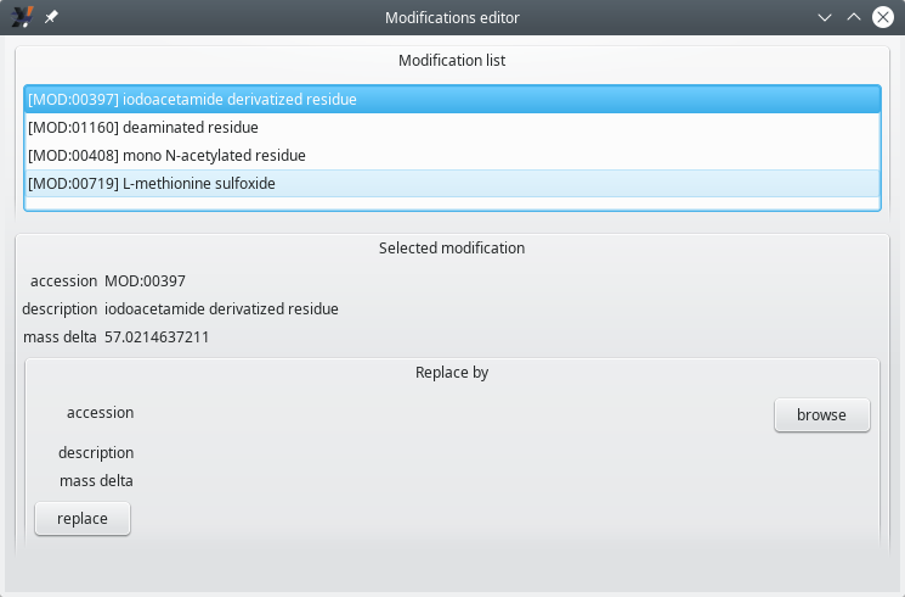
This view shows the details of the modification item currently selected.
Figure 6.3: Modifications editor window (unfolded) #
The Modifications editor window displays the details of the modification currently selected in the Modifications list. The main informational data bits are the following:
accession: the PSI-MOD[15] identificator. This value matches the id field of the corresponding PSI-MOD ontology record (see an example below);
description: the description of the PSI-MOD entity. This value corresponds to the name field of the corresponding PSI-MOD ontology record;
mass delta: the net mass change that results from a protein modification using this modification entity.
Note: A typical term definition from the PSI-MOD ontology
[Term]
id: MOD:00397
name: iodoacetamide derivatized residue
def: "A protein modification that is produced by reaction
with iodoacetamide, usually replacement of a reactive hydrogen
with a methylcarboxamido group."
[PubMed:11327326, PubMed:11510821, PubMed:12422359, Unimod:4]
subset: PSI-MOD-slim
synonym: "Carbamidomethyl" RELATED PSI-MS-label []
synonym: "Iodoacetamide derivative" RELATED Unimod-description []
xref: DiffAvg: "57.05"
xref: DiffFormula: "C 2 H 3 N 1 O 1"
xref: DiffMono: "57.021464"
xref: Formula: "none"
xref: MassAvg: "none"
xref: MassMono: "none"
xref: Origin: "X"
xref: Source: "artifact"
xref: TermSpec: "none"
xref: Unimod: "Unimod:4"
is_a: MOD:00848 ! reagent derivatized residue
If a change, that is, a replacement, is required in the modification, the first step is to click onto the browse button located in the Replace by group box, which opens the PSI MOD selection window shown in Figure 6.4, “PSI MOD selection window”.
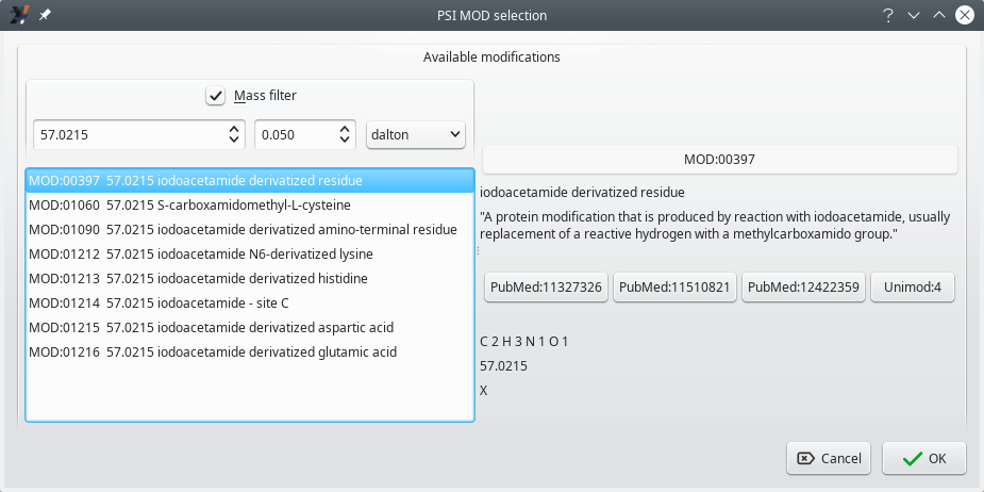
This window allows one to browse the PSI-MOD ontology on the basis of the modification net mass value.
Figure 6.4: PSI MOD selection window #
The window shows the various PSI-MOD-defined ontology terms that match the net mass value of the modification that was selected when the browse button was clicked.
It is possible to search for other PSI-MOD terms that match a different net mass change by modifying the mass value in the spin box widget that is found under the control of the Mass filter check box. The other spin box widget next to the first one allows one to enter the tolerance of the net mass change match. Available “units” for that tolerance are Dalton, ppm and res.
If the Mass filter check box is unchecked, the list beneath it displays all the PSI-MOD ontology terms.
Once the desired term has been found and selected, click the OK button. In order to effectively replace the old term with the new one, do not forget to click the replace button.
Tip: Why bothering with PSI-MOD ontology modification terms ?
It is beneficial to define chemical modifications based on the PSI-MOD ontology terms because the defined modification is then used as its code (for example MOD:00397) in the X!Tandem presets chemical modifications sections (see Section 3.3, “Setting the X!Tandem Run Presets”). Indeed, instead of only entering the net mass, defining a modification preset with the PSI-MOD ontology term code allows i2MassChroQ to compute a number of informative supplementary data. For example, when an isotopic cluster is simulated for a peptide that is modified with a PSI-MOD-documented modification, the formula of the modification recorded in the term is taken into account in the isotopic cluster calculation.
6.2 Configuring Labeling Methods #
i2MassChroQ can handle stable isotope labeling methods. In order to list the available methods, select the menu item of the menu in the main program window. The Labeling methods editor window that opens up is shown in Figure 6.5, “Labeling methods editor window”.
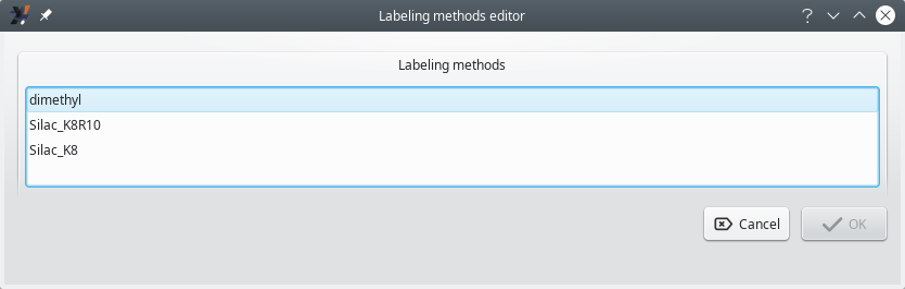
This window lists all the available labeling methods.
Figure 6.5: Labeling methods editor window #
Configuring a labeling method is as easy as selecting one item from the list shown in Figure 6.5, “Labeling methods editor window” and clicking OK. If unsure about the selection, just click Cancel.
Warning: It is not possible to backstep
Once the user has clicked the OK button, it is no more possible to change the labeling method without recomputing the whole data set.
Using a labeling method involves a crucial step during the X!Tandem configuration, as described in Section 3.3, “Setting the X!Tandem Run Presets” and in Caution: Phospho-proteomics projects often involve label-based quantification. Whenever an experiment involves chemical labeling of any protein residue, the scientist is expected to configure the labeling modifications in the X!Tandem presets before running it to search the database, so that the engine models correctly-modified peptides.
When the X!Tandem database search engine has finished identifying peptides and proteins, the modified peptides are only tagged using the modifications' net masses. This is visible in the peptide details window shown in the upper left part of Figure 5.9, “Peptide details window”. There, one can see that the peptide is modified on its lysine 15 residue with one of the labels defined in the Fixed modifications 1 field of the presets configuration window shown in Figure 5.3, “Configuring the phosphorylated residues”.
Selecting a labeling method, as described above, will allow i2MassChroQ to report residue modifications in a much more detailed and informative way. For example, the name of the chemical modification will be used instead of the net mass change due to that modification. For this to happen and the effects to be visible, i2MassChroQ goes back through all the identification process and makes sure that the modified peptides are tagged according to the modifications specified in the labeling method. The effects are visible in the XIC viewer window as shown in Figure 6.6, “Chemical modifications are tagged with their PSI-MOD code ”.
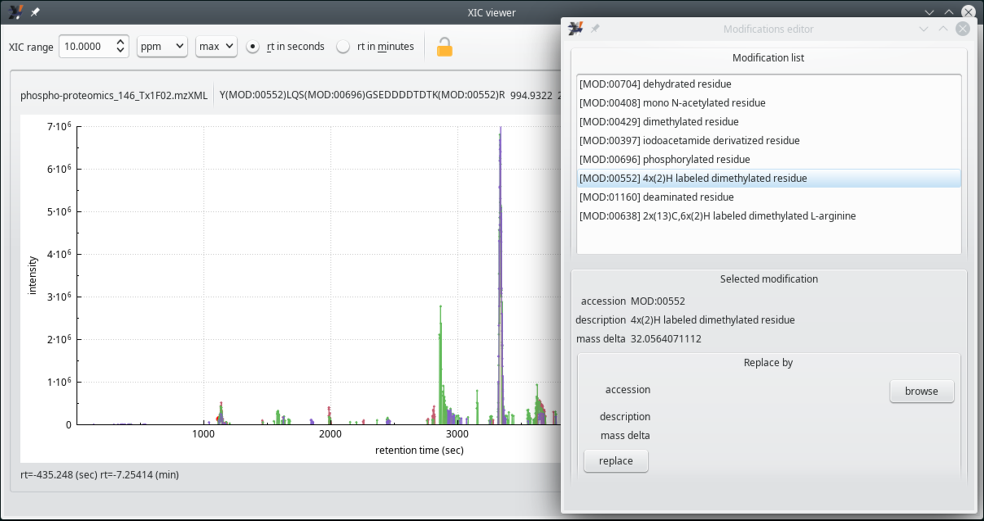
When the user selects a labeling method from the list of available chemical modifications, the chemically modified residue gets an associated tag with the PSI-MOD code. This is visible in this figure, at the top line of the XIC viewer window, where the last peptide's arginine residue is modified with MOD:00552, that corresponds to the 4x(2)H labeled dimethylated residue modification referenced in the Modification editor window on the right hand side of this figure.
Figure 6.6: Chemical modifications are tagged with their PSI-MOD code #
6.3 The i2MassChroQ General Settings #
There are general settings that might be configured for the whole i2MassChroQ program. These settings are available upon selecting the menu of the menu of the main program window. The window that opens up is shown in Figure 6.7, “General settings window”.
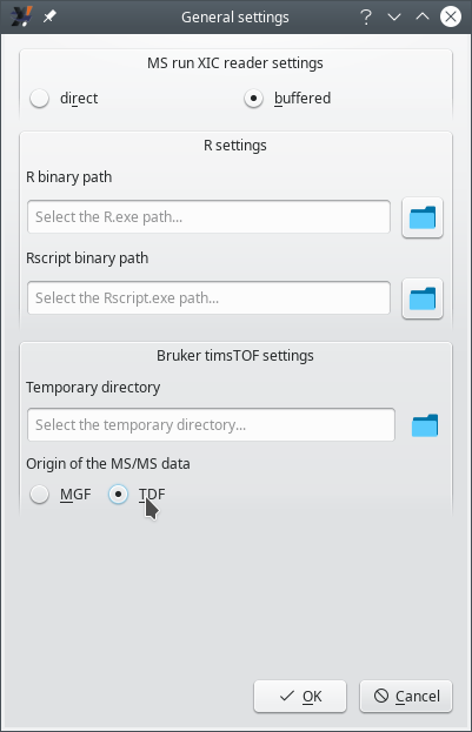
General settings might be configured in this window, as described below.
Figure 6.7: General settings window #
The settings that can be configured are detailed below.
MS run XIC reader settings: if direct is selected, the XIC chromatogram is computed by read the MS data right from the file; if buffered is selected, the XIC chromatogram is computed by using a cache mechanism that buffers data in the computer's memory.
R settings:
R binary path: full path to the R program;
Rscript binary path: full path to the Rscript program;
Bruker timsTOF settings: the fields below need filling-in if the data in the proteomics project originated in the Bruker's timsTOF line of instruments.
Temporary directory: optionally set the full path to the directory where the program stores temporary data;
Origin of the MS/MS data: if MGF is selected, the data are read from the Bruker-generated MGF file; if TDF is selected, the data are read from the Bruker native TDF file.

Warning
If the user selects the MGF option above (because this is what they have), i2MassChroQ will not be able to extract XIC chromatograms, because the MGF file format does not contain MS data, it contains only MS/MS data. If having the native TDF files at hand (in the .d directory), the user is advised to select TDF.
[15] The PSI-MOD ontology is perusable at https://raw.githubusercontent.com/HUPO-PSI/psi-mod-CV/master/PSI-MOD.obo