MassChroQ User Manual
2 Usage
In this chapter, the usage of MassChroQ will be introduced.
2.1 Running MassChroQ #
There are more than one ways to run the MassChroQ software, that will be described in the following sections.
2.1.1 Running MassChroQ from the Terminal #
MassChroQ can be run from the terminal. To get a review of the different options, run the following command at the prompt:
$ masschroq --help
The output of the command above is described in Figure 2.1, “Printing the help message in the terminal”.
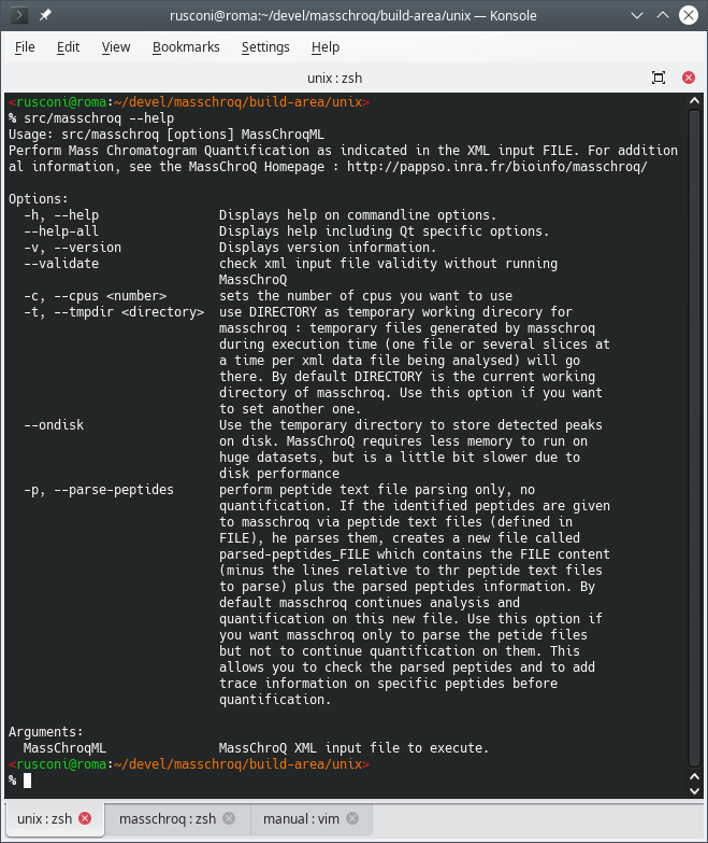
This is the help message that prints out in the terminal when the
program is run with the --help option.
Figure 2.1: Printing the help message in the terminal #
There are two possibilities, either using the
--parse-peptides option or not.
Without the
--parse-peptidesoption. In this case, themasschroqmlfile provided as parameter to the command is fully auto-sufficient: it contains both the MassChroQ configuration bits, the list of identified proteins and the list of identifying peptide precursor ions that need their ion current extracted (XIC).With the
--parse-peptidesoption. In this case, themasschroqmlfile provided as parameter to the command is a partially empty shell: the list of peptide precursor ions is not provided as part of the file. The user must have crafted the peptide list file as described in Figure 1.1, “The peptide list file intsvformat”. The name of that peptide list file must be provided in themasschroqmlfile provided as parameter to the command.
2.1.2 Running MassChroQ from the Graphical User Interface #
MassChroQ can be run from a graphical user interface. To this end, start the following command at the prompt:
$ masschroq_gui
The window that opens up is shown in Figure 2.2, “The main window”.
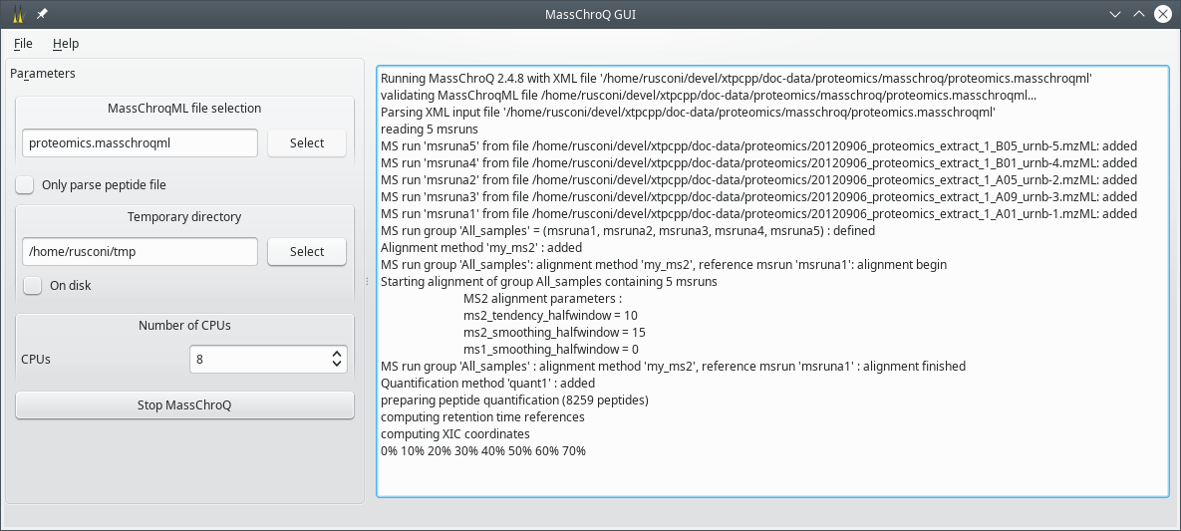
The main window allows the user to easily configure the MassChroQ run. The output of the program in displayed in the text widget on the right hand side of the window.
Figure 2.2: The main window #
The following configuration elements have to be set by the user:
MassChroQML file selection: use the Select button to open a file selection dialog window. Choose the
masschroqmlfile that specifies all the data required to run MassChroQ. This is the input file.Only parse peptide file: check this check box widget if the file set above does not contain the full list of peptide ion precursors that identified the proteins. Checking this widget is analogous to using the
--parse-peptidesoption on the command line (see Section 2.1.1, “Running MassChroQ from the Terminal”).Temporary directory: use the Select button to open a directory selection dialog window. Choose the directory where MassChroQ will write the temporary data it needs to carry over its work.
On disk: check this check box widget to write on disk the detected peaks to alleviate the consumption of random access memory. The program is slower because read/write operations on disk are considerably slower than their equivalent in memory.
Number of CPUs: Enter the number of execution threads that MassChroQ is allowed to use when parallelizing its tasks.
Start MassChroQ: click onto this button to start the program. When the program has started the label of this button changes to Stop MassChroq.
When the program is running, the output is printed in the text widget on the right hand side of the window.
2.2 MassChroq Output File #
The output of MassChroQ is stored in a file named according to the
results_<input_file_name>.ods scheme, where the masschroqml extension is removed. The file is
written inside of the directory that contains the input masschroqmlfile.
The format of the output file is the Libreoffice spreadsheet format (Open Document Spreadsheet, ODS). That file contains three tabs, described below:
MassChroq informations - <id>: the <id> suffix identifies the quantification set. This tab contains the configuration bits of the MassChroQ run.

Note
The <id> suffix is not currently of use, actually. MassChroQ was designed to be able to perform multiple quantification runs with different settings on the same set of associated samples. However, this is not currently available in the X!TandemPipeline interface.
<Sample association group>_proteins: the <Sample association group> prefix identifies the group in which samples were associated. There can be one or more such groups. If there is only one group, it has the default All_samples name. If there are more than one group, then they are named according to the configuration set by the user. This tab contains the peptide, protein and protein_description columns, all self-explanatory.
peptides_<id>_<Sample association group>: the <id> and <Sample association group> suffixes were described above. This tab contains the actual XIC data for each peptide precursor ion that was quantified. There are a large number of columns, described below.
The peptides tab in the results spreadsheet file contains a large number of columns that are described in the paragraphs below. Each row of the spreadsheet is related to a given peptide precursor ion that was fragmented and that allowed identifying a peptide, which in turn contributed to the identification of a protein.
quantification: this is the <id> mentioned above. At the time of writing this identification bit is not significant.
group: this is the name of the group of associated samples.
msrun: automatically-assigned name that is unambiguously attached to the mass spectrometry file.
msrunfile: mass spectrometry data file name for the current sample.
mz: calculated (FIXME) m/z value.
rt: retention time at which the peptide precursor ion was eluted (in seconds). FIXME (the rt for the most intense ion current, that is, the top of the curve or what ? see rtbegin below)
maxintensity: maximum ion current intensity. FIXME
area: are under the XIC chromatogram curve. FIXME
peak quality: assessment on the quality of the peak. The more 'a' characters, the higher the quality. FIXME
rt begin: the retention time at the beginning of the the precursor ion XIC chromatogram peak. FIXME c'est vrai ?
rt end: the retention time at the end of the the precursor ion XIC chromatogram peak. FIXME c'est vrai ?
peptide: the peptide id as set by the dabase search engine.
label: label that might be attached to the peptide in labelling-based quantification. FIXME
sequence: the sequence of the peptide precursor ion.
z: the charge of the peptide percursor ion.
mods: the chemical modifications encountered on the peptide. The format is like [C 37 H 55 O 10 N 10 S 0], which is the net elemental composition of the modifications FIXME !!!!!
ninumber: the isotopologue (0 for monoisotopic, 1 for 1 heavy isotope, 2 for 2 heavy isotopes).
nirank: rank of the peak in the previous column in the isotopic cluster. FIXME
niratio: ratio
:
:
:
:
:
:
:
:
:
:
: